
Criança portadora de fenilcetonúria. Demonstração que a vida, mesmo com limites, manifesta esta alegria.
A fenilcetonúria é caracterizada por uma doença genética recessiva hereditária, relacionada ao cromossomo autossômico 12, com freqüência atingindo aproximadamente uma criança a cada 12 mil nascimentos, variando de acordo com a população, acometendo em maior número indivíduos de pele clara, sendo muito rara em africanos.
O organismo portador, devido homozigose do alelo alterado para esse gene, expressa má formação ou mesmo ausência da enzima fenilalanina hidroxilase, que cataliza a conversão do aminoácido fenilalanina em tirosina, a partir do acréscimo de grupamento hidroxila (OH).
Essa deficiência provoca distúrbios na síntese de proteínas, causada pela carência de tirosina e acúmulo de fenilalanina no organismo.
Entre as implicações da concentração de fenilalanina, estão relacionados fatores de toxicidade devido à transformação deste aminoácido em: ácido fenil-pirúvico e ácido fenil-lático, substâncias tóxicas que provocam lesões nas células do sistema nervoso central, causando atraso no desenvolvimento psicomotor (locomoção e comunicação), hiperatividade e convulsões.
Causas

A fenilcetonúria é hereditária, isto é, passa de pais para filhos. O pai e a mãe devem passar o gene defeituoso para que o bebê tenha essa doença. Isso é conhecido como traço recessivo autossômico.
Os bebês com PKU não possuem uma enzima chamada fenilalanina hidroxilase, necessária para quebrar um aminoácido essencial denominado fenilalanina. Essa substância é encontrada em alimentos que contêm proteínas.
Sem essa enzima, os níveis de fenilalanina e de duas substâncias associadas a ela crescem no organismo. Tais substâncias são prejudiciais ao sistema nervoso central e causam dano cerebral.
Exames

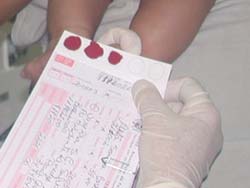
A fenilcetonúria pode ser fácilmente detectada em um simples exame de sangue. Em todos os estados brasileiros, o exame de triagem para PKU (ou fenilcetonúria), chamado teste do pezinho, é exigido para todos os recém-nascidos como parte do painel de triagem. O teste normalmente é realizado por meio da retirada de algumas gotas de sangue do bebê antes da saída dele do hospital.
Tratamento
O tratamento mais eficiente para fenilcetonúria é uma dieta especial, que ajuda a controlar a quantidade de fenilalanina consumida (alguma fenilalanina é necessária para o crescimento e desenvolvimento). Pessoas com fenilcetonúria que seguem essa dieta a partir do nascimento, ou logo depois, se desenvolvem normalmente e freqüentemente não apresentam sintomas.
Nenhum comentário:
Postar um comentário